第二部 【企業情報】
第1 【企業の概況】
1 【主要な経営指標等の推移】
(注) 1.当社は連結財務諸表を作成しておりませんので、連結会計年度に係る主要な経営指標等の推移については記載しておりません。
2.「収益認識に関する会計基準」(企業会計基準第29号 2020年3月31日)等を第24期の期首から適用しており、第24期から第27期に係る主要な経営指標等については、当該会計基準等を適用した後の指標等となっております。
3.持分法を適用した場合の投資利益については、当社は関連会社を有していないため記載しておりません。
4.1株当たり配当額及び配当性向については、配当を実施していないため記載しておりません。
5.潜在株式調整後1株当たり当期純利益については、潜在株式は存在するものの、当社株式は非上場であり、期中平均株価が把握できないため、また、1株当たり当期純損失であるため記載しておりません。
6.第23期から第27期までの経常損失及び当期純損失の計上は、医療用医薬品の開発のための研究開発活動に係る先行投資によるものであります。
7.自己資本利益率については、当期純損失であるため記載しておりません。
8.株価収益率は、当社株式が非上場であるため記載しておりません。
9.第23期、第24期及び第25期については、キャッシュ・フロー計算書を作成しておりませんので、キャッシュ・フローに係る項目については記載しておりません。
10.第26期及び第27期の財務諸表については、「財務諸表等の用語、様式及び作成方法に関する規則」(昭和38年大蔵省令第59号)に基づき作成しており、金融商品取引法第193条の2第1項の規定に基づき、RSM清和監査法人の監査を受けております。なお、第23期、第24期及び第25期については、「会社計算規則」(平成18年法務省令第13号)の規定に基づき算定した各数値を記載しており、当該監査を受けておりません。
11.従業員数の(外書)は、臨時従業員の年間雇用平均人員数であります。臨時従業員には派遣社員を含めております。
12.2025年4月1日付で普通株式1株につき普通株式500株の割合で株式分割を行っております。さらに、2025年6月13日付で第三者割当増資(A種種類株式1,442,200株の発行)を行い、資本金は748,990千円となり、2025年6月20日付で第三者割当増資(普通株式33,300株の発行)を行い、資本金は763,975千円となりましたが、2025年12月6日付で無償減資を行い、資本金は100,000千円となりました。
2026年1月13日付で普通株式を対価とする取得条項に基づき、発行済種類株式の全てを当社が取得し、引き換えに種類株主に対して当社普通株式の交付を行い、同日付で当社が取得した種類株式の全てを消却しております。なお、当社は、2026年1月9日開催の臨時株主総会決議に基づき、2026年1月14日付で、種類株式を発行する旨の定款の規定を廃止しております。本書提出日現在の発行済株式総数(普通株式)は、6,828,000株となっております。
第26期の期首に当該株式分割が行われたと仮定し、1株当たり純資産額及び1株当たり当期純損失を算定しております。
13.2025年4月1日付で普通株式1株につき普通株式500株の割合で株式分割を行っております。東京証券取引所自主規制法人(現 日本取引所自主規制法人)の引受担当者宛通知「『新規上場申請のための有価証券報告書(Ⅰの部)』の作成上の留意点について」(2012年8月21日付東証上審第133号)に基づき、第23期の期首に当該株式分割が行われたと仮定して算定した場合の1株当たり指標の推移を参考までに掲げると、以下の通りとなります。
なお、第23期、第24期及び第25期(1株当たり配当額についてはすべての数値)については、RSM清和監査法人の監査を受けておりません。
2 【沿革】
松本 正(当社設立者)は、1989年から協和醱酵工業株式会社(現:協和キリン株式会社)において、海外バイオベンチャーの有望な新薬プログラムを探索し、共同開発プログラムを立ち上げる業務に従事していました。その中で、将来の医薬品開発をリードしていくのはバイオベンチャーであることを確信し、バイオベンチャーと積極的に提携し、新しい医薬品を生み出す仕組みが必要であると考えていました。そして、1997年秋に、“患者さんに求められる医薬品のよりスピーディな開発を支援すること”を目的として、社内ベンチャー制度に応募し、第1号社内ベンチャーに認定されました。
3 【事業の内容】
(1) 事業の概要
当社は設立以来”Required Medicine for all who request it”(人々に求められる医薬品)の開発に取り組んでおります。
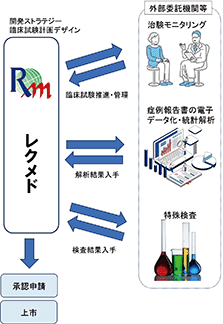
具体的には、患者の皆様に寄り添い現場のニーズを汲み取りながら、国内外のベンチャー企業やアカデミアとの強力な研究ネットワークを生かし、患者の皆様が必要としているアンメット・メディカル・ニーズ(※1)の薬剤シーズを見つけ出します。そして、当社が主体となって研究開発戦略を練り、CRO(※2)等を有効活用しながら医薬品の臨床試験(※3)に必要な基礎データを取得します。さらに、その薬剤の特性に合わせた臨床試験を自らデザインし、ベンチャー企業という小さな組織でありながらも、自らが治験依頼者として、規制当局(本邦においては、独立行政法人医薬品医療機器総合機構)との相談・合意を得て、新薬の製造・販売承認申請に必要な第Ⅲ相試験まで自社で行っております。

当社は、すでに2剤の新薬を上市させることができました。そして今、NaPPSの変形性膝関節を対象とした第Ⅲ相臨床試験を実施しています。

ⅰ アンメット・メディカル・ニーズに特化した医薬品開発
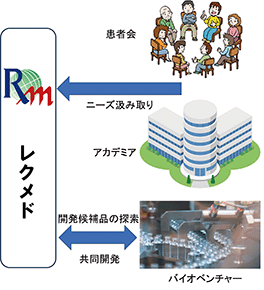
③国立研究開発法人新エネルギー・産業技術総合開発機構(以下「NEDO」という)による「中堅・中小企業への橋渡し研究開発促進事業」としての選定を受け、ムコ多糖症(※5)患者を対象としたポリ硫酸ペントサンナトリウムの安全性・有効性にかかる(医師主導の)臨床研究を国立大学法人岐阜大学(現 国立大学法人東海国立大学機構 岐阜大学)と共同で行いました。
更に、「医療上の必要性の高い未承認薬・適応外薬検討会議」(厚生労働省)の開発要請品目にも複数応募し、患者数が少ないがゆえに有効な治療が確立されず、薬剤貢献度が低い疾患の治療薬や、従来の治療に対する患者満足度が低い疾患に対して新たな治療薬を届けるべく、これらの疾患の開発品を探索してきました。そして、有望な開発候補品を保有している国内外のバイオベンチャー企業等と共同で医薬品開発プログラムを立ち上げ、当社の開発ノウハウを駆使して患者対象の臨床研究を実施し、薬理効果を実証することで、開発候補品の価値を更に高めております。具体的には、「医療上の必要性の高い未承認薬・適応外薬検討会議」(厚生労働省)の開発要請品目の
①ベタイン原末(適応症:ホモシスチン尿症)
②コール酸(適応症:先天性胆汁酸代謝異常症)
の2品目を開発して、製造販売承認を得てこれらの薬剤を必要としている患者に届けることができました。バイオベンチャーとの共同開発としては、PsychoGenics, Inc.(現:PGI Drug Discovery LLC)と共同開発を進め、既知物資であるエルトプラジンに対して注意欠陥多動症や進行期パーキンソン患者におけるL-dopa誘発ジスキネジアに対する新規効能を見出し、米国における注意欠陥多動症の第Ⅱ相試験、欧州におけるパーキンソン病のL-dopa誘発ジスキネジア(※6)に対する第Ⅱ相試験、さらに米国在住日本人を対象とした薬物動態試験を実施しました。
ⅱ 自社による医薬品開発・製造販売承認
の体制をとります。卸売業者へ販売する販売業者が病院や薬局への情報提供を担いますが、①患者数がごく少数に限られる領域では、開発担当者自らが医療従事者と密接にコンタクトを取り、その製品の開発で培った疾患・製品知識と臨床試験の際に関わった医療従事者とのコネクションを生かしたプロモーション活動を行うことにより、より専門的かつ効果的な販売推進を展開できます。一方で、②患者数の多い領域では、強力な販売ネットワーク・組織基盤を有する他社との提携による販売推進を展開します。このような販売体制をとることで、収益の最大化を目指すと共に同時に企業価値が最大になるような事業戦略を取っていきます。
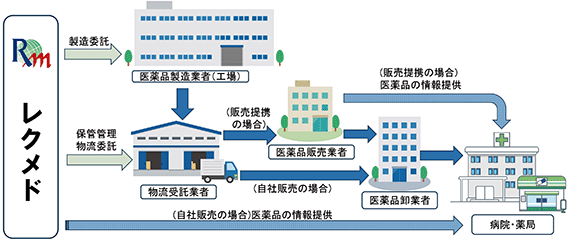
上記「自社販売」と「販売提携」の区分けは、製品の適応症ごとに、下表のような患者数と施設数に自社基準を設けて振り分けています。
ⅲ コンサルティング事業
長年の国内外のバイオベンチャーとの共同開発や医薬品開発のノウハウが社内に蓄積しており、それらを活用したい国内外のバイオベンチャーやアカデミアからの依頼により、業務提携サポートや創薬戦略立案の支援等を行っております。
(2) 当社の収益構造
当社の事業セグメントは医薬品事業の単一セグメントであります。
医薬品開発プロセスにおける当社の事業領域、事業系統図及び売上の種類は以下のとおりです。
(医薬品開発プロセスにおける当社の事業領域)
医薬品の開発は一般的には以下のプロセスを経て進められ、同プロセスにおける当社の各事業の対象領域は以下のとおりです。
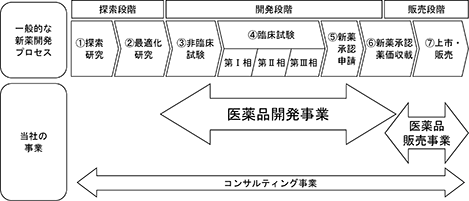
① 探索研究(疾患の治療ターゲットの確認とターゲットに作用する化合物や抗体などの探索)
② 最適化研究(探索研究で得られた化合物や抗体の最適化)
③ 非臨床試験(臨床試験を開始するために必要な動物試験)
④ 臨床試験(ヒトでの有効性と安全性を評価する試験)
⑤ 新薬承認申請(厚生労働省あるいは各国又は地域の医薬品許認可審査機関への製造販売承認申請)、照会事項対応
⑥ 新薬承認、薬価収載
⑦ 上市・販売(医療機関関係者・患者の皆様への提供)
(事業系統図)
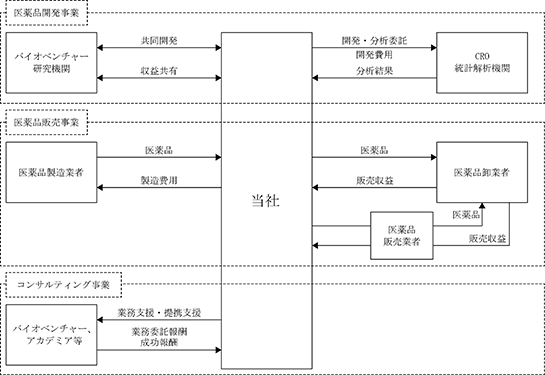
(売上の種類)
当社の売上は以下のもので構成されております。
<医薬品販売事業:上市済み医薬品>
サイスタダン原末(一般名:ベタイン)はホモシスチン尿症の治療薬で、厚生労働省「医療上の必要性の高い未承認薬・適応外薬検討会議」での審議を経て国内企業の募集が行われたことに対して当社が応募し、その後さらに希少疾病用医薬品(※7)として指定され、国内医薬品として開発し、製造・販売の承認を得たものです。輸入元であるRecordati Rare Diseases SARLから製品を輸入し2014年より自社販売を行っておりましたが、10年間の契約満了に伴い2022年1月1日にRecordati Rare Diseases SARLの日本法人レコルダティ・レア・ディジーズ・ジャパン株式会社に承継しました。
オファコルカプセル50mg(一般名:コール酸)は先天性胆汁酸代謝異常症(※8)の治療薬です。こちらも、厚生労働省「医療上の必要性の高い未承認薬・適応外薬検討会議」での審議を経て国内企業の募集が行われたことに対して当社が応募し、その後さらに希少疾病用医薬品として指定され、国内医薬品として開発し、製造・販売の承認を得たものです。
先天性胆汁酸代謝異常症は、肝臓においてコレステロールから胆汁の成分であるコール酸(オファコルカプセル50mgの有効成分)やケノデオキシコール酸を生合成する酵素に先天的な遺伝子変異で異常が起こり、生合成が途中で止まってしまう疾患です。生合成が途中で止まると有害な異常代謝物が肝臓に有害な作用を示し、肝障害を誘発します。また、胆汁酸が不足し、食事から必要な脂溶性成分(ビタミンA、ビタミンDを含む)を吸収できなくなり、種々の成長障害を引き起こします。
先天性胆汁酸代謝異常症は、障害を受けた酵素の種類により、12種類の病型に分かれています。現在オファコルカプセル50mgの治療が行われているのは3種類の病型で、その一つに40名近い患者数の報告がなされている脳腱黄色腫症(※9)があります。これは胆汁酸生合成酵素のうち、ステロール27位水酸化酵素をコードする(生成情報を有する)遺伝子CYP27Aが欠損し同酵素の活性が低下する病型で、体内(特に脳や腱)に徐々にコレステロールやコレスタノール(コレステロールの類似物質)が蓄積し、精神発達遅滞・認知機能障害・錐体路症状(運動麻痺)・小脳症状などの進行性神経障害、アキレス腱黄色腫及び若年性白内障、早発性心血管疾患などを発症します。また、新生児~乳児期の遷延性黄疸・胆汁うっ滞や若年発症の慢性の下痢で発症する症例も報告されています。新生児期や小児期に発見されるケースはこれまで少なく、本疾患に対する早期診断と早期治療開始が大きな問題となっていました。
脳腱黄色腫症以外の病型では、3β-hydroxy-Δ5-C27-steroid dehydrogenase (3β-HSD)欠損症と3-oxo-Δ4-steroid 5β-reductase (Δ4-3-oxoR)欠損症の2病型が知られています。こちらは合わせて国内に10名程度の患者がいて、新生児期の黄疸や胆汁うっ滞の症状で発見されます。こちらの病型に関しては、新生児のスクリーニングが積極的に行われつつあり、2024年に1名の新生児が先天性胆汁酸代謝異常症(Δ4-3-oxoR 欠損症)と診断され、オファコルカプセル50mgの治療を開始しました。早期診断により早くオファコルカプセル50mgによる治療を開始して進行を押さえ症状の改善を図り、結果的にオファコルカプセル50mgの売上が伸びていくことを期待しております。
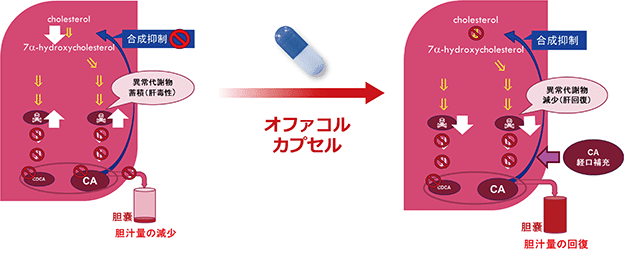
肝臓ではコレステロールを出発物質として、胆汁の成分であるコール酸(CA)とケノデオキシコール(CDCA)という2つの胆汁酸が合成され胆嚢(たんのう)の中に蓄えられます。食物を摂取すると胆嚢からこれら2つの胆汁酸が腸の中に分泌し、脂肪成分と一緒になって腸から体内に取り込まれます。もし、先天性胆汁酸異常症に代表される症状、すなわち胆汁酸が合成されず食物摂取と同時に分泌されない状態になると脂肪成分(脂溶性ビタミンも含む)が体内に取り込まれず、成長不良等の障害が発生します。幸いなことにこの胆汁酸は、食事と一緒に薬として飲むことができます(CA補充療法)。食事中の脂肪成分は、一緒に飲んだコール酸(CA)と一緒に体内に取り込まれ、脂肪成分と切り離された後に今度は胆嚢に溜まって、次の食事の時の胆汁酸としてリサイクルできるようになります。
オファコルカプセル50mgは1カプセルの中に50mgのコール酸が有効成分として含まれています。このコール酸は肝臓においてコレステロールを出発物質として生合成される胆汁酸の重要な成分の一つで、食事をした後に胆嚢から十二指腸に分泌され、食事中の脂質(含む脂溶性ビタミン)の腸からの吸収を助けます。先天性胆汁酸代謝異常症は、このコール酸の生合成に係る酵素異常によって、コール酸が生合成されず、毒性の高い代謝中間体が肝臓に蓄積し、肝障害を発生させる疾患です。また、このコール酸を生合成する経路において、最終産物のコール酸によって生合成の調整が行われますが、コール酸ができないことで「コール酸を作れ」という指令を止めることができず、毒性の高い代謝中間体が蓄積する悪循環が発生します。
食事と共に服用されるオファコルカプセル50mgの中に含まれているコール酸は、あたかも胆嚢から十二指腸に分泌されるコール酸と同じようにふるまい、腸において脂質(脂溶性ビタミンを含む)と共に吸収され、肝臓に再吸収
の適応に関しては、藤本製薬株式会社のフジケノン粒状錠125mg(一般名はケノデオキシコール酸)が2025年9月に承認されました。脳腱黄色腫症は、以前よりケノデオキシコール酸の治療が行われており、フジケノン粒状錠125mgは脳腱黄色腫症の患者を対象に、藤本製薬株式会社が第Ⅲ相試験を実施したものです。フジケノン粒状錠125mgは脳腱黄色腫症治療の第一選択薬として認められていますが、一部患者に対し安全性の面から第一選択薬だけでは十分量を確保できず、オファコルカプセル50mgが追加投与されると期待しております。
(3) 医薬品開発事業としての当社の開発パイプライン
当社は後期臨床ステージ(通常第Ⅱ相試験以降)に相当する開発物質を複数保有しております。
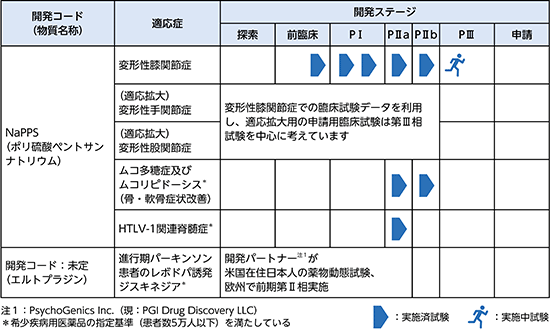
① ポリ硫酸ペントサンナトリウム
ポリ硫酸ペントサンナトリウム(下図)はブナから抽出された植物由来の半合成物質で、欧州で1949年に抗血栓症治療薬として開発されるなど古くから知られており、1996年には米国で間質性膀胱炎治療薬として、2017年には欧州で膀胱痛症候群の治療薬として承認・販売されています。また、動物用医薬品としては犬の骨関節炎治療薬として日本や欧米各国で販売されており、オーストラリアでは犬と馬の関節炎治療薬として販売されています。
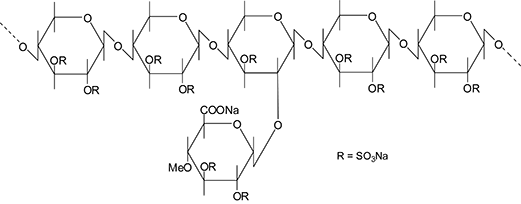
小麦を含め広く植物界に存在するペントサン(d-キシロースが重合した化合物の総称)、およびその硫酸化化合物であるポリ硫酸ペントサンナトリウムは第二次世界大戦後のドイツにおいて当時供給量が少なかったヘパリン(抗血液凝固剤)の代用品として、bene pharmaChem GmbH & Co. KG.(以下「bene社」という。)が研究を開始し、1949年にドイツで承認され、その後他ヨーロッパ諸国に広まりました。この時、bene社は製造方法をノウハウとして社内に留めるためにあえて特許を出願しなかったので、その製法は現在に至るまでbene社内で管理されています。このためbene社が製造するポリ硫酸ペントサンナトリウムに匹敵するものの製造は至難の業となっています。bene社はポリ硫酸ペントサンナトリウムの製造において特殊なノウハウを有しており、それは技術移管を行っても同じ分子量領域の製品を忠実に再現できないほど難しいものです。そのため、bene社から高品質のポリ硫酸ペントサンナトリウムを輸入することは医薬品の品質・有効性・安全性の観点から極めて重要であり、当社ビジネスの競争優位性となっています。当社は2004年から日本国内における開発で、bene社との共同開発を進めております。さらに、bene社の品質の高いポリ硫酸ペントサンナトリウムに加え、薬剤の長期の安定性を付与することを目的に、当社独自で凍結乾燥製剤を開発し、特許を国際出願しました。既に、日本、オーストラリア、欧州で特許を取得し、米国は審査中です。
当社以外のポリ硫酸ペントサンナトリウムの注射剤は液剤です。液剤では長期保存中、バイアル瓶の内側で注射液とガラス内面が常に接触した状態となり、ガラスの表面に付着していた微小のガラスの膜が剝げ落ちることがあります。このようなことが起きた場合、その製品ロット全体を回収しなければならなくなるリスクがあります。一方、凍結乾燥製剤は、水分が完全に蒸発した状態でありガラス表面との反応がありませんので、長期間安定する製剤となり、薬剤の保存も室温保存が可能になります。
ポリ硫酸ペントサンナトリウムの作用としては、各種の慢性炎症を抑え、生体機能を正常化します。変形性関節症では関節組織への複合的作用により関節症の症状を緩和し、ムコ多糖症ではグリコサミノグリカン(※11)による全身の慢性炎症を抑えながら骨・軟骨症状(特に関節の拘縮)を抑えます。また、成人T細胞白血病リンパ腫の原因ウイルス(HTLV-1)が感染したT細胞の脊髄内浸潤を抑制し、HAMの下肢機能障害を改善する効果を期待しております。
日本以外の展開では、2010年9月にbene社と共同で本薬剤の韓国における開発及び販売権をChong Kun Dang Pharmaceutical Corp.(以下「CKD社」という。)に与えるライセンス契約を締結しております。このライセンス契約は、bene社との共同開発の一環として、当社が韓国における開発パートナー探しを行いました。CKD社はbene社よりポリ硫酸ペントサンナトリウム原薬を輸入し、間質性膀胱炎治療薬として販売しております。CKD社に対しては、bene社と共同でライセンス契約を結び、このライセンス契約のベースとなった当社とbene社の共同開発契約では、あらゆる利益は50:50で折半するとなっていることから、この韓国における原薬売上総利益の一部を当社が受領でき、CKD社がbene社へ支払った原薬売上代金の一部をbene社から当社が受領し、売上を計上しております。会計上はbene社への売上高として計上されますが、当社からbene社へ製品やサービスを提供した対価ではありません。
・変形性膝関節症
変形性膝関節症診療ガイドライン2023(日本整形外科学会)の序文には、「日本は老年人口比率が2007年に21%を超え,今や3割に達しようとする超高齢社会となっています。超高齢社会の到来は疾病構造を変化させ,運動器疾患では運動器障害による移動機能低下-ロコモティブシンドロームに日本は直面しています。その主たる疾患の一つである変形性膝関節症は,約800万人が疼痛や硬さ,腫れなど何らかの症状を有しており,X線学的な関節症変化は約2500万人に存在し,40歳以上で有病率が約55%,有症状者が1800万人に達するといわれ,要介護への移行リスクが約6倍あることが指摘されています。変形性膝関節症は個人の日常生活のみならず,社会の経済活動や医療・福祉政策に多大な影響を与えており,その克服は日本の喫緊の課題といえます。」と記載されています。
2005年から2007年にかけて東京大学 吉村典子 特任教授らが実施した大規模住民コホートROAD STUDYが実施され、少なくとも一つの関節がKellgren and Lawrence分類(※10)(以下、「KL分類」という。)Grade2以上ありと診断された変形性膝関節症の有病率は、年齢が高くなるにつれて上昇しています。(出典:N. Yoshimura et al, J Bone Miner Metab 27:620-628 (2009))。
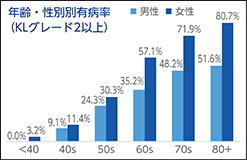
変形性関節症の中でも変形性膝関節症は、加齢、肥満、遺伝的因子、力学的負荷など多くの原因によって発症し、膝関節軟骨の表層に近い部位から軟骨細胞外基質(マトリックス)の消失、軟骨の菲薄化、亀裂形成、軟骨細胞のクラスター形成や細胞死、関節周縁部の骨棘形成の変化と進行する疾患です。
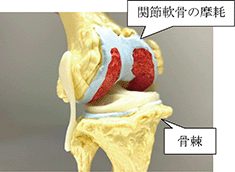
療薬を目指しております。今までの関節痛を抑える鎮痛剤とは異なり、疾患修飾剤(※12)として新しい薬剤に分類されます。そのために、これまでの薬剤ではできなかった変形性膝関節症の進行の抑制を期待しています。さらに、本薬剤は鎮痛剤やヒアルロン酸製剤等の既存の薬剤と競合するものではなく併用できるので、幅広い患者層に処方が可能になると考えております。また、皮下注射製剤であることから、整形外科医のみならず、その他の診療科の医師や看護師、更には将来の自己注射も可能な薬剤に発展させることが可能な薬剤となっています。
これまでの非臨床試験(動物実験)において、ポリ硫酸ペントサンナトリウム皮下注射において膝関節の炎症や軟骨の変性を抑える効果が示されております。また、軟骨オリゴマーマトリックスタンパク質(以下「COMP」)という、コラーゲンネットワークを安定化させ軟骨組織の構造を維持する構造タンパク質があります。変形性膝関節症により軟骨が損傷または分解されるとCOMPが放出され、その濃度が上昇することから、COMPは軟骨代謝のバイオマーカーとして、疾患活動性、関節損傷の進行、および治療効果のモニタリングに利用されます。当社の前期第Ⅱ祖試験および後期第Ⅱ相試験の2つの臨床試験において、このCOMPの量の減少がみられ、ポリ硫酸ペントサンナトリウムが変形性膝関節症における膝軟骨の破壊を抑制していることが示唆されています。現時点までの試験結果を基に、当社では変形性膝関節症の進行に伴う症状と、それに対するポリ硫酸ペントサンナトリウムの想定されている作用を以下の通りに考えています。
当社では、第Ⅲ相試験に入る前の最終の探索的臨床研究と位置付けられている後期第Ⅱ相試験において、KL分類Grade 2~4の変形性膝関節症患者様を対象被験者とし、WOMAC総合指数(※14)の変化量及び変化率を主要評価項目として、目標症例数90例(実薬群:プラセボ群=1:1、実薬群に対しては2mg/kgのポリ硫酸ペントサンナトリウムを週1回皮下注射で8週間投与し、プラセボ群に対してはポリ硫酸ペントサンナトリウムではなく生理食塩水を投与)にて比較することを計画し、2022~2024年に実施しました。この治験において、被験者の組入れの際に、KL分類Gradeの群間の偏りで臨床効果に差が出る恐れがあることや、KL分類 Grade 4の患者ではポリ硫酸ペントサンナトリウムの薬理効果発現までに期間を要し、限られた評価期間では明確な効果が示されない可能性も考えられたので、試験結果の解析の中でKL分類 Grade 2&3と4に分けて層別解析を行えるよう、それぞれにおいてポリ硫酸ペントサン群とプラセボ群で同数になるように動的割付(※15)を行って治験を実施しました。膝等の変形性関節症に関連する症状を評価するWOMAC総合指数は、被験者に点数を付けてもらうことからプラセボ効果の影響を受けやすいと懸念されたため、仮登録時のWOMAC-Aの値は5以上9以下、また1年以上前から治療を行っているにも関わらず痛みがある、さらに、医師が実際に膝を押して痛みがあることを確認する等、対象被験者の治験への登録基準を通常の試験よりも厳格化しました。その結果、被験者の確保に苦戦し、複数回の登録期間の延長を行いましたが、無理な被験者エントリーは有効性評価のノイズを増加させるリスクが高まること、更にこの治験のために製造した治験薬の使用期限が2024年1月となっており、本登録後14週間の治験薬投与期間を確保するため、最終的に被験者の仮登録を2023年9月に締め切りました。その結果、同意取得88例、仮登録58例、本登録49例となりました。2024年3月に、本登録された全ての被験者の最終来院が完了し、その後集積されたデータの解析を実施しました。
その結果、KL分類Grade4を含む全ての被験者(実薬群25例、プラセボ群23例)では、後期第Ⅱ相試験の主要評価項目であった、WOMAC総合指数で、NaPPS投与群はプラセボ投与群に比べて改善傾向を示したものの、両群間に明確な差異は認められませんでした。一方、後期第Ⅱ相試験開始前の対面助言時に、医薬品医療機器総合機構より製造販売承認申請時の評価は、WOMACスコアのうち特に痛みに重点を置いた評価項目であるWOMAC-Aでの有効性の証明が必要であるとの追加情報を得ており、WOMAC-Aを評価しました。このWOMAC-Aの軽減に関し、ポリ硫酸ペントサンナトリウム投薬群(実薬群)がプラセボ群との比較において、KL分類Grade4を含む全ての被験者では、t-検定(2つのグループの平均値に統計的な差があるかどうかを検定する統計学的手法)で統計学的に有意な差を認めることに至りませんでしたが、KL分類 Grade 2&3の層別解析(実薬群12例、プラセボ群11例)の結果では、実薬群がプラセボ群と比較して、t-検定において統計学的に有意な差を以って、痛みを軽減していることが示されました。
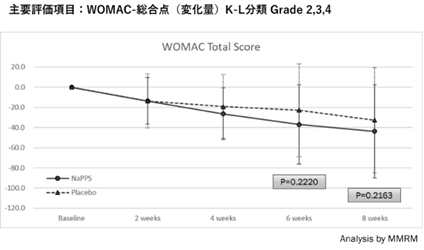
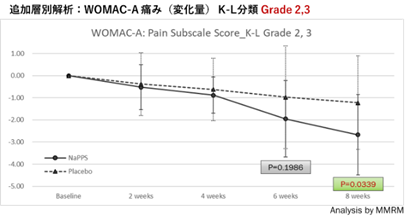
以下の2つのグラフは、KL分類2&3の個々の被験者の治験薬の投与開始後(V3)の痛みの変化を表しています。
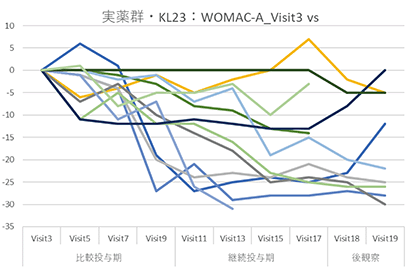
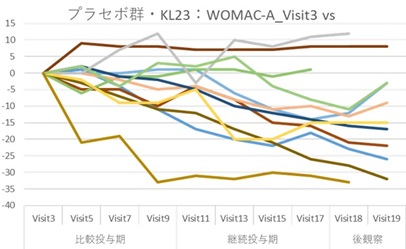
上記の結果、KL分類Grade2&3の患者を対象とすることで、第Ⅲ相試験を行うに足る有効性を確認できたと当社は判断し、第Ⅲ相試験の治験届を2024年12月27日に提出しました。その概要は、下表の通りです。
全体のスケジュールは下記の通りです。
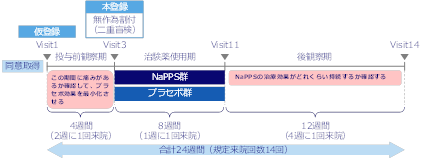
また、国内製造受託会社へ委託して治験薬(実薬とプラセボ薬)を製造し、2025年7月24日に最初の被験者への投与を開始し(本登録)、2026年2月12日時点で、本登録を済ませた被験者数が53名となりました。
ポリ硫酸ペントサンナトリウムを変形性膝関節症治療薬として上市した場合のサプライチェーンは、
①bene社よりポリ硫酸ペントサンナトリウムの原薬を輸入します。
②治験薬を製造した医薬品受託製造会社へ商用製品の製造を委託します。
③2025年に販売基本契約を締結した帝國製薬株式会社に商用製品を販売します。
④帝國製薬株式会社から医薬品と卸業者を通して、全国の医療機関・薬局に商用製品を届けます。
サイスタダン原末やオファコルカプセル50mgに比べて変形性膝関節症の患者数は桁違いに大きく、今後当社の収益構造の拡大に大きく寄与し、企業価値を高める上で最重要プログラムと位置付けております。更に、ポリ硫酸ペントサンナトリウムは皮下注射により全身に作用し、個々の関節に注射することなく全ての関節への作用が期待できることと、ポリ硫酸ペントサンナトリウムが既存の変形性膝関節症治療と投与方法や作用機序で競合関係にないことから、多くの患者さんに利用頂けると考えております。
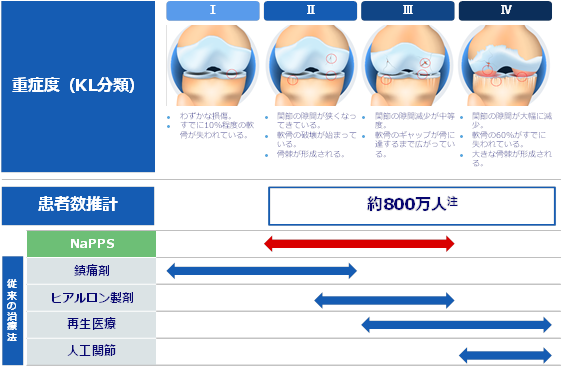
注 日本整形外科学会「変形性膝関節症診療ガイドライン2023」
更に、皮下注射により薬剤を血流に乗せて全身に届けられることから、変形性膝関節症のみならず変形性手関節症や変形性股関節症に対しても適応拡大が期待できると考えており、これらに対し適応を拡大することも計画しております。
・変形性手関節症
変形性手関節症は、(特に人差し指と中指)の指先と真ん中の関節や親指の根元の関節に変形性関節症が起きる疾患で、手の痛みだけでなく、手の機能低下や巧緻な運動が障害されて日常運動や生活の質の低下を引き起こします。2022年に発表された「科学研究費助成事業の研究成果報告書(課題番号:18K17374)」において、国内で初めての疫学研究がなされ、重度の変形性手関節症の発生率は7.5/1000人・年、増悪率が8.6/1000人とされています。国内人口を約1億2000万人とすると、年間約70万人の新規患者が発生し、年間約100万人の患者さんの症状が悪化していると推定しております。しかしながら、膝関節と違い手関節は関節が小さく、局所投与の薬剤は使いにくく、変形性膝関節症の治療で多用されているヒアルロン酸は変形性手関節症に対して適応症を持っていないのが現状です。これに対し、ポリ硫酸ペントサンナトリウムは、皮下投与で全身に薬剤の効果が期待できるので、変形性手関節に対して新しい治療選択肢をもたらすことができると考えております。当社では、ポリ硫酸ペントサンナトリウムの変形性膝関節症の第Ⅲ相試験が終了後、速やかに本適用の適応拡大を目指した第Ⅲ相試験の実施を計画しています。
・変形性股関節症
変形性股関節症は、足の付け根にある“股関節”の軟骨が徐々にすり減り、歩行などの動作時に痛みを引き起こす病気で、症状の進行に伴い日常生活に影響を及ぼし、生活の質の低下を引き起こします。重症化すると股関節を人工関節に置換する施術が行われ、日本人工関節学会のTHAレジストリー2022によると2022年度に約8万件の人工股関節置換手術が行われています。また、変形性股関節症の有病率1%(出典:日本整形外科学会診療ガイドライン委員会、変形性股関節症診療ガイドライン策定委員会「変形性股関節症診療ガイドライン」)を日本の人口に換算して120万人の潜在患者数が存在すると推定され、ヒアルロン酸製剤としては、ジクロフェナクエタルヒアルロン酸ナトリウム(商品名:ジョイクル)が変形性股関節症に対して適応症を取得していますが、同剤投与により重篤なショック、アナフィラキシーが発現する恐れもあり、変形性股関節症は当社が目指すアンメットな疾患の一つと考えられます。当社では、上記変形性手関節症同様に、ポリ硫酸ペントサンナトリウムの変形性膝関節症の第Ⅲ相試験が終了後速やかに本適用の適応拡大を目指した第Ⅲ相試験の実施を計画しています。
・ムコ多糖症及びムコリピドーシス(※16)
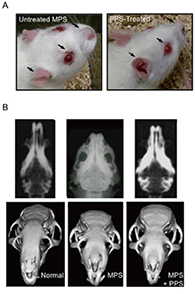
また、これまでの治験を通して、ムコ多糖症患者の一部は変形性関節症を併発していることが判明しています。このことから、ポリ硫酸ペントサンナトリウムが変形性膝関節症の適応で承認を受けた場合、ムコ多糖症の一部の患者については変形性膝関節症(合併症)治療の目的でポリ硫酸ペントサンナトリウムを処方できる可能性が生じます。それ以外の小児を含めた患者が治療を受けられるよう、変形性膝関節症の試験終了後に、適応拡大を目的とした第Ⅲ相試験(場合によっては、市販後臨床試験の形式で実施)の実施を検討しております。
・HTLV-1関連脊髄症(HAM)(※17)
HAMは、成人T細胞白血病・リンパ腫の原因ウイルスであるHTLV-1感染によって惹き起される進行性の脊髄炎であり、歩行障害、排尿障害等を伴います。2014年の日本医療研究開発機構研究班の調査では、全国のHTLV-1感染者(キャリア)数は72~82万人と推定されています。また、別の報告では、HAMの推定有病率は人口10万人中3人、患者数は3,600人弱と推定されています(出典:厚生労働科学研究費補助金(新型インフルエンザ等新興・再興感染症研究事業)「本邦におけるHTLV-1感染及び関連疾患の実態調査と総合対策」平成21年度総括研究報告書)。治療は、ステロイドやインターフェロンαが用いられていますが、ステロイドは長期投与による他の感染症の誘発、糖尿病の悪化、骨粗鬆症による骨折などの副作用、インターフェロンαではうつ症状や肝障害、白血球減少などの副作用が知られており、新規の治療法が求められています。
当社ではJSTの研究成果最適展開支援プログラムを活用し、国立大学法人長崎大学と共同でポリ硫酸ペントサンナトリウムの新規HAM治療薬開発に向けた(医師主導の)検証的臨床研究を実施しました。その結果、ポリ硫酸ペントサンナトリウムはHTLV-1感染T細胞の脊髄内浸潤を抑制し、脊髄内の慢性炎症を抑え、下肢機能障害の改善を示すことを確認できました。この結果を元に「HTLV-1関連脊髄症の予防または治療剤」の特許を2012年に出願し、日本、米国、英国、ドイツで特許が成立しています。その後、NEDOの支援を受け、国立大学法人長崎大病院においてHAM患者を対象とした(企業主導の)探索的臨床試験を実施しました。これらの結果を元に、後期第Ⅱ相試験以降の臨床試験を変形性膝関節症の第Ⅲ相試験終了後に開始する計画です。
② エルトプラジン
エルトプラジンはセロトニン5HT1A/1B(※18)のパーシャルアゴニスト(※19)で、中枢性疾患の動物モデルで注意欠陥・多動性障害の他、パーキンソン病におけるレボドパ誘発性ジスキネジア(LID)での有効性が確認されております。当社は、2003年にPsychoGenics Inc.(現:PGI Drug Discovery LLC)との間で本剤の共同開発を開始し、米国において成人の注意欠陥・多動症の第Ⅱ相試験を実施し、更に米国在住の日本人と欧米人との間の薬物動態比較試験を実施することで、日本を始めとするアジア地区における注意欠陥・多動性障害を適応症とする開発を進め、最終的に他社へライセンス供与する戦略を描いていました。しかし、ライセンス先に開発を委ねる戦略は、相手先の戦略変更によって容易に開発スケジュールの変更が行われ得ることから、申請まで自社で開発し、薬剤の市場性に応じて自社販売するか販売提携を行うかの選択を行って製品価値を最大化する戦略に変更し、2019年に共同開発契約を終了して最適な開発戦略を模索してきました。
その結果、注意欠陥・多動症の開発は中断し、当社の強みである希少疾病用医薬品としてパーキンソン病の後期に現れるレボドパ(L-Dopa)(※20)服用時のジスキネジア等の適応症に対して独自に開発を進めることとしました。LIDは、希少疾病用医薬品の対象疾患(国内患者数が5万人以下)となり得ることから、同適応症の治療薬を目指すことを決断し、2025年にLIDの国内での臨床試験開始に向けてPGI Drug Discovery LLCとData Sharing and Licensing Agreementを締結し、同社が持つデータの提供を受けながら準備を進めております。
既にPsychoGenics Inc.(現:PGI Drug Discovery LLC)との共同開発期間中に実施した米国在住の日本人と欧米人との間で薬物動態に相違がみられないことを示した試験(国内の第Ⅰ相試験に相当)や、欧州でパーキンソン患者を対象とした第Ⅱ相試験が実施済みであることから、国内の開発においては第I相試験をスキップして第Ⅱ相試験から臨床試験を進められるメリットがあると考えております。現在、当社では国内で治験薬に用いるエルトプラジン原薬の製造を委託するべく候補企業と協議を進めており、今後GMP基準を満たした治験薬の原薬を製造し、その治験薬を用いた国内第Ⅱ相試験開始に向けて準備を進めております。
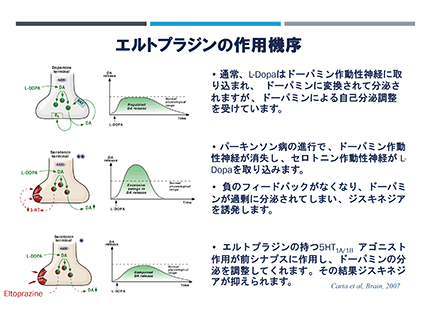
パを取り込んでドーパミンに変換して脳内でドーパミンを放出します。しかし、セロトニン作動性神経細胞はドーパミンの脳内濃度を調整する機能がなく、脳内でドーパミンの濃度が一気に上昇してしまい、この時に患者が自分で運動をコントロールできず、ジスキネジアが発生してしまいます。エルトプラジンは、このセロトニン作動性神経細胞に働きかけ、ドーパミンの急激な放出を抑えることで、ジスキネジアの発生を抑えることを可能にする薬剤です。
(4)当社の知的財産
当社の開発品のポリ硫酸ペントサンナトリウムは、1949年にドイツで製品化された物質で、開発者があえて特許を出願しなかったことによりヨーロッパブナの木からペントサンを抽出し、硫酸基を導入して製造する工程が製造ノウハウとして保護されています。
当社では、このポリ硫酸ペントサンナトリウムに対し以下の特許を取得しています。特に、①の製材特許は安定性に優れたポリ硫酸ペントサンナトリウムの凍結乾燥製剤を製造する特許で、室温にて3年以上の安定性を確立しています。
4 【関係会社の状況】
該当事項はありません。
5 【従業員の状況】
(1) 提出会社の状況
(注) 1.従業員数は就業人員数であります。
2.従業員数の(外書)は、臨時従業員の年間雇用平均人員数であります。
3.臨時従業員には派遣社員を含めております。
4.平均年間給与は、賞与及び基準外賃金を含んでおります。
5.当社は単一セグメントであるため、セグメント別の従業員数を記載しておりません。
(2) 労働組合の状況
労働組合は結成されておりませんが、労使関係は円満に推移しております。
(3) 管理職に占める女性労働者の割合、男性労働者の育児休業取得率及び労働者の男女の賃金差異
当社は「女性の職業生活における活躍の推進に関する法律(平成27年法律第64号)」及び「育児休業、介護休業等育児又は家族介護を行う労働者の福祉に関する法律(平成3年法律第76号)」の規定による公表義務の対象ではない為、記載を省略しております。