第二部 【企業情報】
第1 【企業の概況】
1 【主要な経営指標等の推移】
(注) 1.当社は連結財務諸表を作成しておりませんので、連結会計年度に係る主要な経営指標等の推移については記載しておりません。
2.第15期は販売費及び一般管理費を上回る補助金収入を営業外収益に計上したため、当期純利益を計上しております。
3.持分法を適用した場合の投資利益については、関連会社がないため記載しておりません。
4.当社は、2020年2月28日付で第三者割当増資(C種優先株式31,664株の発行)を行い、資本金は574,960千円となり、2020年4月3日付で第三者割当増資(C種優先株式24,668株の発行)を行い、資本金は944,980千円となりましたが、2020年7月30日付で無償減資を行い、資本金は100,000千円となりました。さらに、2020年8月31日付で第三者割当増資(普通株式5,000株の発行)を行い、資本金は300,000千円となり、発行済株式総数は、普通株式57,053株、A種優先株式61,000株、B種優先株式8,000株、C種優先株式56,332株となりましたが、2020年9月23日付で普通株式を対価とする取得条項に基づき、発行済優先株式の全てを当社が取得し、引き換えに優先株主に対して当社普通株式の交付を行い、同日付で当社が取得した優先株式の全てを消却しております。なお、当社は、2020年9月16日開催の臨時株主総会決議に基づき、2020年9月23日付で、種類株式を発行する旨の定款の規定を廃止しております。加えて、2020年11月12日付で普通株式1株につき20株の株式分割を行ったため、本書提出日現在の発行済株式総数(普通株式)は、3,647,700株となっております。
5.1株当たり配当額及び配当性向については、配当を実施していないため記載しておりません。
6.潜在株式調整後1株当たり当期純利益については、第14期、第17期及び第18期におきましては、潜在株式は存在するものの、当社株式は非上場であり、期中平均株価が把握できないため、また、1株当たり当期純損失であるため、第15期におきましては、潜在株式は存在するものの、当社株式は非上場であり、期中平均株価が把握できないため、第16期におきましては、1株当たり当期純損失であり、また、潜在株式が存在しないため記載しておりません。
7.自己資本利益率については、第14期は当期純損失であり債務超過であるため、第15期は債務超過であるため、第16期は当期純損失であり期首において債務超過であるため、第17期及び第18期は当期純損失であるため、記載しておりません。
8.株価収益率は当社株式が非上場であるため記載しておりません。
9.第14期、第15期及び第16期についてはキャッシュ・フロー計算書を作成しておりませんので、キャッシュ・フローに係る各項目については記載しておりません。
10.従業員数は就業人員(当社から社外への出向者を除き、社外から当社への出向者を含む。)であり、臨時雇用者数は従業員数の100分の10未満であるため、記載しておりません。
11.第14期、第15期及び第16期については、「会社計算規則」(平成18年法務省令第13号)の規定に基づき算出した各数値を記載しており、当該各数値については、金融商品取引法第193条の2第1項の規定に基づくEY新日本有限責任監査法人による監査を受けておりません。
12.第17期及び第18期の財務諸表については、「財務諸表等の用語、様式及び作成方法に関する規則」(昭和38年大蔵省令第59号)に基づき作成しており、金融商品取引法第193条の2第1項の規定に基づき、EY新日本有限責任監査法人により監査を受けております。
13.2020年11月12日付で普通株式1株につき20株の株式分割を行っており、第17期の期首に当該株式分割が行われたと仮定し、1株当たり純資産額及び1株当たり当期純損失を算定しております。
14.2020年11月12日付で普通株式1株につき20株の株式分割を行っております。
そこで、東京証券取引所自主規制法人(現 日本取引所自主規制法人)の引受担当者宛通知「『新規上場申請のための有価証券報告書(Ⅰの部)』の作成上の留意点について」(平成24年8月21日付東証上審第133号)に基づき、第14期の期首に当該株式分割が行われたと仮定して算定した場合の1株当たり指標の推移を参考までに掲げると、以下のとおりとなります。
なお、第14期、第15期及び第16期の数値(1株当たり配当額については全ての数値)については、EY新日本有限責任監査法人の監査を受けておりません。
2 【沿革】
(注)KP-100ITはKP-100を主成分とする、脊髄腔内投与用製剤です。
KP-100LIはKP-100を主成分とする、局所投与用製剤です。
3 【事業の内容】
(1) 概要(背景)
当社は、難治性疾患、すなわち「症例数が少なく、原因不明で、治療法が確立しておらず、生活面への長期にわたる支障がある疾患」に対する治療薬の研究開発を目指す大学発バイオベンチャーとして設立されました。
設立後、中村敏一氏(当時:大阪大学大学院医学系研究科教授)の発見したHGF(Hepatocyte Growth Factor:肝細胞増殖因子)タンパク質を開発パイプライン*として導入し、組換えDNA技術*を応用したタンパク質(以下、「組換えタンパク質」という)の製造法の確立、非臨床試験*の実施を経て、欧米及び日本における臨床試験*を複数実施いたしました。

(2) 事業モデル
当社が想定している事業モデルを図2に示します。当社は対象疾患や提携先に応じてA、B、Cを組み合わせたハイブリッド型の事業モデルを志向しております。その中でも、臨床試験の成果をより確実に医薬品として社会実装するため、自社での医薬品製造販売承認申請を行うことを基本方針とします。当社の臨床段階のパイプラインについては、脊髄損傷急性期と声帯瘢痕はAとBのハイブリッド(自社開発と販売提携)、ALSと急性腎障害はBによる事業化を目指しております。また、クラリス・バイオセラピューティクス社への原薬供給はCに該当します。
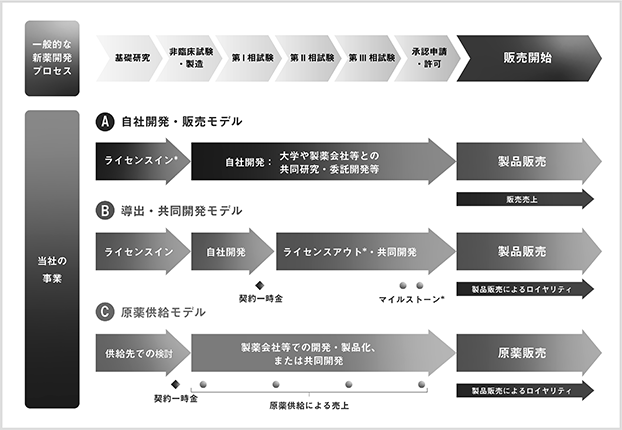
<図2:当社の事業モデル>
また、開発については一般的な新薬開発プロセスに従って実施する必要がありますが、難治性疾患に対する治療薬の開発を実現するために以下に示すようなプロセスを実施しております。
① 基礎研究*
医薬品の候補となるシーズ*探索は長期の研究期間が必要である上に、この段階で候補が見つかっても医薬品として成功する確率は非常に低いことがわかっております。当社では、基礎研究として、組換えヒトHGFタンパク質の新規適応症の探索及び新規候補品の探索を行っておりますが、大学との共同研究において専門的な知識を活用することにより、成功確率を高めるよう基礎研究を進めております。
② 非臨床試験・製造
非臨床試験や製造については、開発業務受託機関、製造受託機関等の専門技術を活用することにより、迅速に進めております。また、この開発段階では膨大な支出に対する収入が得られないため、公的資金(助成金、補助金)を活用することにより、自社負担を減らし、経済的なリスクを低減させております。
③ 臨床試験(第I相試験*、第Ⅱ相試験*、第Ⅲ相試験*)
患者数の少ない難治性疾患では、疾患の原因も病態も明らかでないことがあり、臨床試験の評価項目の設定や症例の選定等が難しいとされております。当社では、大学との連携により、難治性疾患に対する専門的な知見を集積し、小規模で成功確度が高い評価項目を策定し臨床試験を実施しております。臨床試験の一部の業務については、開発業務受託機関に委託して品質・開発速度を確保するとともに、臨床評価や解析については、専門病院及び大学病院の医師と連携して科学的な質を高めております。また、大学病院等が公的資金を確保し、臨床試験(医師主導治験)の実施を希望する場合には、治験薬及び科学的な情報・知識を提供して治験推進に協力し、成果の独占的な利用許諾を得ます。
④ 承認申請・許可
臨床試験の成果をより確実に医薬品として社会実装するため、自社での医薬品製造販売承認申請を行うことを基本方針とします。難治性疾患を対象として開発しているため、POCが得られた後には厚生労働省の「希少疾病用医薬品指定」を受け、開発費用の助成、優先審査の活用などにより申請までの業務を加速することが可能です。また、「条件付き早期承認制度」「先駆的医薬品指定制度(旧 先駆け審査指定制度)」等の制度を活用することにより申請・審査に係る時間を短縮し、早期承認を目指します。
⑤ 販売
難治性疾患は高度医療機関において治療が行われるため、開発した医薬品の取り扱い施設の数が限定されます。したがいまして、通常の医薬品のような大規模な流通販売網の構築が不要となります。また、競合する医薬品が少ないことから営業活動に大きな費用をかける必要がありません。当社が医薬品製造販売業の許可を取得し、医薬品卸売販売業者と提携することで、必要な場所に必要な医薬品を届けるサプライチェーンの構築が可能です。その結果、販売に係るコストが削減され、売上総利益を高い割合で継続的に得られるメリットを享受することができます。
なお、脊髄損傷急性期を対象とした製品のサプライチェーンについては、現時点で当社内に営業販売体制がないことから、下図に示すように丸石製薬株式会社と東邦ホールディングス株式会社との提携により構築されております。丸石製薬株式会社は救急領域のスペシャリティファーマ*として国内の救急病院をカバーする営業体制を有しているため、当該製品の販売及びプロモーションの提携をすることで、サプライチェーンを構築することが可能であると考えております。
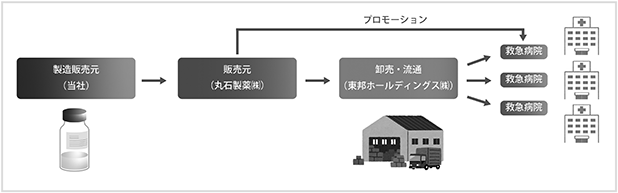
<図3:脊髄損傷急性期を対象とした製品のサプライチェーン>
⑥ 適応拡大
個々の難治性疾患は患者数が限られますが、複数パイプラインの開発を進めること、海外へ販売を拡大すること、他領域の疾患についても開発を進めることにより、市場の拡大が可能であると考えております(図4)。例えば、脊髄損傷急性期及びALSにおいてHGFの神経保護作用が示された場合には、脳神経領域の疾患に対しても神経保護作用を示し、治療薬として開発される可能性が考えられます。また、声帯瘢痕においてHGFの抗線維化作用が示されると、声帯以外の線維化が原因となる疾患への応用が考えられます。さらに、急性腎障害において静脈内投与でのHGFの効果が確認された場合には、腎臓以外の臓器における難治性疾患への応用の可能性が考えられます。HGFは様々な作用を持っていることから、一つの作用についての効果が確認されると、他領域への応用の可能性があると考えております。将来的には、当社において適応拡大による市場の拡大を行いながら他の製薬企業から他領域での開発を希望する提案を受けた場合には、原薬を供給することにより、継続的な収益を得ることも可能であると想定しております。
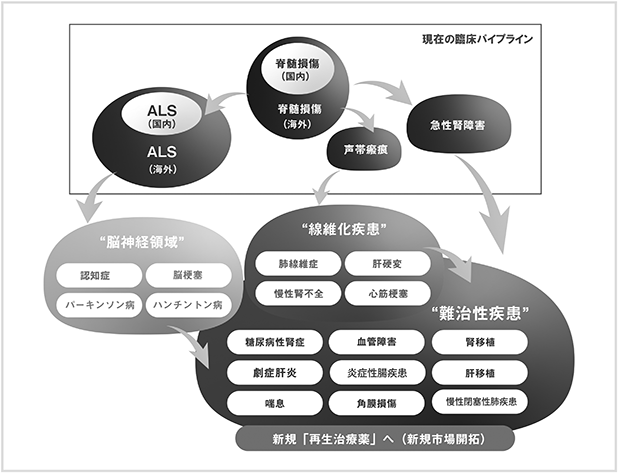
<図4:適応拡大の可能性>
なお、当社の事業セグメントは、医薬品開発事業の単一セグメントであり、事業系統図(図5)及び収益の種類(表1)は以下のとおりであります。
(事業系統図)
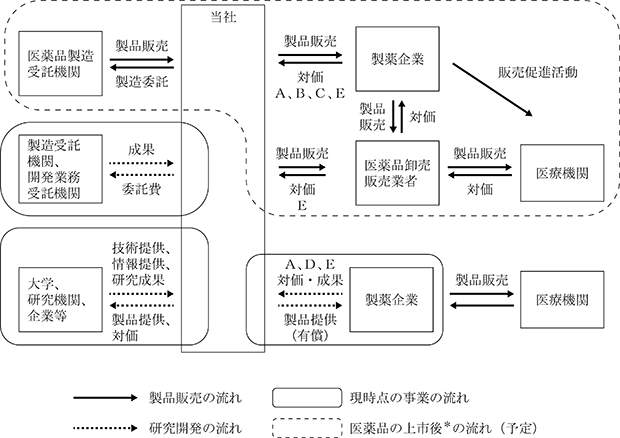
<図5:事業系統図>
<表1:収益の種類>
(3) 創薬シーズ
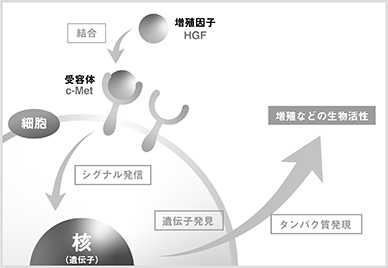
HGFはその後の研究によって、細胞増殖以外にも細胞保護、形態形成等の生物活性*を併せ持つことが明らかになり、対象となる細胞も肝細胞だけでなく、腎臓、肺、皮膚等の細胞に対して効果があることが示されました(図7)。特に、線維成分の蓄積により細胞の機能が低下する「線維化」や「硬化」を解除する作用(抗線維化という)及び神経細胞・グリア細胞*等の神経系細胞に対する生物活性が明らかになると、複数の難治性疾患に対する治療薬の候補として様々な研究成果が報告されました。
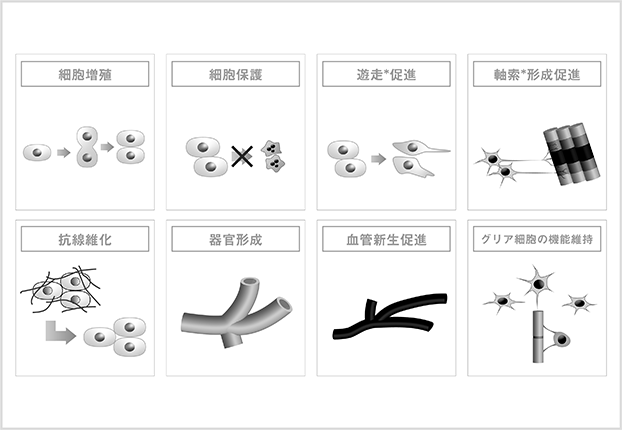
<図7:HGFの生物活性>
実験動物を用いて疾患モデルを作成すると、体内のHGFタンパク質量が上昇し、疾患が治癒することが示され、HGFと組織修復の関係が明らかになってきました。そこで、組換えヒトHGFタンパク質を疾患モデルに補充的に投与すると症状の軽減や治療効果が示されました。組換えヒトHGFタンパク質を用いた研究は現在でも多くの疾患領域について行われておりますが、臨床応用の可能性がある代表的な疾患は下表のようになります。
<表2:組換えヒトHGFタンパク質の臨床応用の可能性がある疾患>
※太字は当社が現在医薬品の開発をしている疾患
当社は、HGFの発見者である中村敏一氏(当時:大阪大学大学院医学系研究科教授)より2005年に組換えヒトHGFタンパク質の開発実施権の許諾を受け、難治性疾患を対象としたパイプラインとして開発を開始いたしました。
(4) 製造体制
組換えヒトHGFタンパク質を製品(医薬品)として製造するためには、①原薬製造及び②製剤製造の大きく2種類に分けられる製造を行うことが必要です。この2種類の製造についてはいずれも当社が実施権を保有しており、製造受託会社に委託して製品の製造を行っております。医薬品の製造については、「医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律」(以下、「薬機法」という)及び「医薬品及び医薬部外品の製造管理及び品質管理の基準に関する省令」(以下、「GMP*省令」という)に基づき、厳格な品質管理の下で製造を行う必要があります。
また、組換えタンパク質を医薬品として製造する場合には細胞を用いた製造法になるため、複雑で特別な技術が多数必要となります(図8)。当社は、組換えヒトHGFタンパク質の製造方法(原薬製造及び製剤製造)のノウハウを有しており、2社の製造受託会社に原薬製造及び製剤製造を各々委託することにより、製品の供給を行っております。

<図8 製造工程の概略>
開発段階では、製品販売による収入が得られず、定期的な製造計画も決められないため、自社で製造設備を有して人材を確保・管理するよりも外部の受託機関を活用する方が経済的なリスクが低く、効率的と考えております。また、難治性疾患を対象としているため、必要となる製品量も小スケールであることが想定されます。したがいまして、開発から販売初期までは現在の製造体制を持続いたしますが、適応症の追加や拡大等、開発状況に応じてスケールアップ検討を進める予定です。さらに、現在米国においてドラッグマスターファイル*の登録を完了しており、提携会社が米国において組換えヒトHGFタンパク質の開発を行う場合には、当社の製造法を提携会社に開示することなく当社が提携会社に原薬販売することが可能となる体制を構築しております。
(5) 製剤開発
HGFは発見から長い年月が経過しているため、物質特許はすでに失効しておりますが、当社は医薬品の製剤として設計・開発する過程で2種類の製剤組成について特許を取得しております。1つは長期的な安定性を目的とした凍結乾燥製剤(特許1)、もう1つは長期的な安定性に加え、神経疾患の治療に適用できるよう組成を改良した凍結乾燥製剤(特許2)です。
<表3:製剤組成に関する特許>
(2020年10月31日現在)
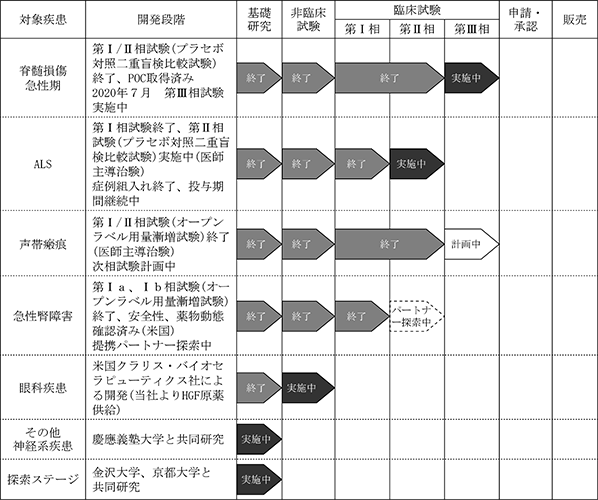
(6) 開発の状況
開発の状況を図9に示します。臨床試験までステージが進んでいるパイプラインは4件(脊髄損傷急性期、ALS、声帯瘢痕、急性腎障害)、動物疾患モデルにおいて有効性が認められ、臨床試験準備のステージに進んでいるパイプラインが1件(眼科疾患)、基礎研究のステージにあるパイプラインが複数あります。現在は最も開発ステージの進んでいる脊髄損傷急性期を対象とした医薬品開発に注力し、製造販売承認を得ることにリソースを集約しております。一方、基礎研究については大学等との共同研究を継続し、新規適応症の開拓、新規シーズの探索を行っております。
<図9:パイプラインの開発状況>
(2020年10月31日現在)
① 脊髄損傷急性期を対象とした開発
脊髄とは脳と体をつなぐ神経が集積している組織であり、脊椎と呼ばれる骨の中に保護されております。脊髄損傷とは、事故や転倒により脊髄に強い外力が加わり、組織が損傷を受けた結果、運動神経や感覚神経の機能が失われ、運動障害や感覚障害を発症する疾患です。国内では年間約5千人の新規患者が発生すると報告されております(坂井宏旭ら、Journal of Spine Research、2010年)。しかしながらこれまでのところ、損傷した脊髄に対する有効な治療法はなく、脊髄を囲む脊椎の骨折や脱臼を治療するための手術や、リハビリテーションによる残存神経機能の有効利用と日常生活動作の獲得など、対症療法のみとなっております。
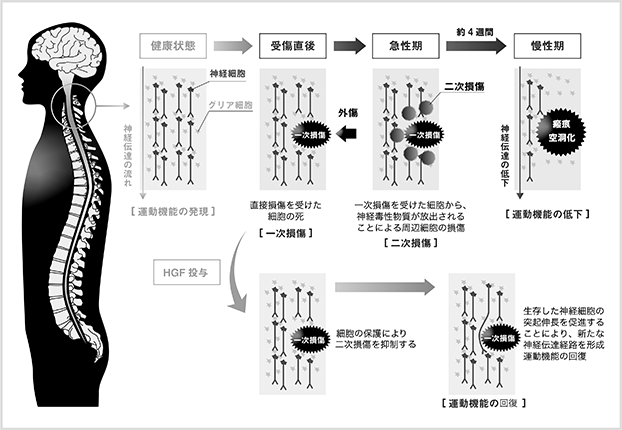
<図10:脊髄損傷の発症メカニズムとHGFによる治療効果>
HGFは神経細胞に対し保護作用を示すこと、軸索伸展*の促進作用があることから、脊髄損傷の治療薬として開発される可能性があると考えられます。脊髄損傷は外力によって引き起こされる組織の損傷(一次損傷という)に続いて周辺組織に損傷が広がる二次損傷が起こります(図10)。脊髄損傷の急性期においては、この二次損傷を抑えることが治療につながると考えられております。当社では慶應義塾大学医学部整形外科との共同研究により、脊髄損傷モデル動物を用いて組換えヒトHGFタンパク質の薬理効果を確認する試験を実施したところ、運動機能評価において有効性が確認されました。そこで、医薬品の開発に必要な非臨床試験(毒性試験、薬物動態*試験など)を実施して、臨床試験に開発ステージを進めました。脊髄損傷急性期患者を対象とした第Ⅰ/Ⅱ相試験結果の概要を表4に示します。
<表4:脊髄損傷急性期患者を対象とした第Ⅰ/Ⅱ相試験結果の概要>
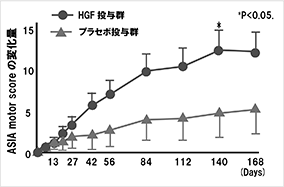
本試験の結果を踏まえて、HGF(脊髄腔内投与用製剤:KP-100IT)は2019年9月に厚生労働省により脊髄損傷急性期を対象とした希少疾病用医薬品指定を受けました。また、本試験の結果は、国際医学雑誌Journal of Neurotraumaにも論文発表されております(オンライン公開:2020年5月22日、DOI: 10.1089/neu.2019.6854)。この試験で得られたPOCを検証する目的で次の第Ⅲ相試験の計画を策定し、下表に示す概要で2020年7月より第Ⅲ相試験を開始しました。
<表5:脊髄損傷急性期患者を対象とした第Ⅲ相試験計画の概要(実施中)>
第Ⅲ相試験は2022年後半に終了予定であり、有効性を示すデータが得られましたら製造販売承認申請を進める予定としております。
② ALS
ALSは「難病の患者に対する医療等に関する法律」において難病指定を受けている難治性神経疾患の1つで、運動神経細胞が選択的に障害を受けるため、筋力の低下、筋肉の萎縮が引き起こされる疾患です。その結果、歩行困難、言語障害、嚥下障害及び呼吸障害等の症状が進行的に現れ、発症後3~5年で約80%の患者が死亡すると言われております。国内患者数は約1万人(平成30年度特定医療費(指定難病)受給者証所持者数、難病情報センター)と報告されております。すでに承認された医薬品が2剤ありますが、いずれも効果は限定的であり、より効果の高い新規治療薬の開発が強く望まれています。実際の医療現場では、リハビリテーションによる生活動作の維持・獲得、鎮痛剤を用いた痛みの抑制などの対症療法が既存治療法となっております。
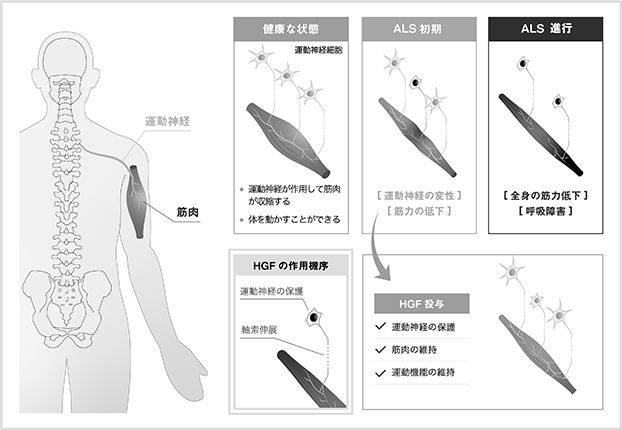
<図12:ALSの発症メカニズムとHGFの治療効果>
ALSの発症要因は遺伝によるもの、グルタミン酸毒性によるもの、原因不明のものと様々ですが、運動神経細胞が障害を受け脱落することにより筋肉の萎縮が起こることが共通する現象であるため、運動神経細胞を保護することが治療効果につながると考えられます。したがって、前項の脊髄損傷同様に、HGFの神経細胞に対する保護作用、軸索伸展の促進作用はALSの治療につながる可能性があると考えられます(図12)。当社では、東北大学医学部脳神経内科との共同研究により、ALSモデル動物を用いて組換えヒトHGFタンパク質の薬理効果を確認する試験を実施したところ、運動機能の回復、発症の遅延、生存期間の延長といった効果が確認されました。そこで、医薬品の開発に必要な非臨床試験(毒性試験及び薬物動態試験など)を実施して、臨床試験に開発ステージを進めました。ALS患者を対象とした第Ⅰ相試験結果の概要を下表に示します。
<表6:ALS患者を対象とした第Ⅰ相試験結果の概要>
当該試験の主要評価項目である安全性及び忍容性の確認について、重篤な副作用は認められず、良好であることが示されました。また、副次評価項目として、適切な投与量を策定するために薬物動態を検討した結果、有効性を検討するために必要な投与量についての知見が得られました。この試験で得られた結果を基に、POCの確認を目的とした第Ⅱ相試験を実施しております。ALS患者を対象とした第Ⅱ相試験の概要は下表のとおりです。
<表7:ALS第Ⅱ相試験計画の概要(実施中(患者組入れ終了))>
当該試験は医師主導治験であるため、当社は治験薬提供者として携わり、試験終了後の事業化に関しては当社が独占的に行う契約となっております。2021年後半には開鍵*の予定であり、POCが得られたと判断された場合には条件付き早期承認制度及び希少疾病用医薬品指定制度の申請に適するかどうかを検討し、第Ⅲ相試験の実施要否とともに製造販売承認申請への戦略を迅速に策定していく予定です。
③ 声帯瘢痕
声帯瘢痕とは、声帯の物性が固く変化(線維化、瘢痕化)して動きが悪くなるため、声が出しにくくなる音声障害を生じる疾患です。発症原因は明らかになっていませんが、声帯の外傷や炎症、声帯の手術後などに起こりやすいことが知られております。患者数については、小規模の疫学調査結果(平成21年度厚生労働科学研究費補助金(難治性疾患克服研究事業)「声帯溝症の診断治療の確立と、標準化に向けたガイドラインの作成に関する研究」)から、国内に3,000~12,000人の患者がいると推定されております。これまでのところ、声帯瘢痕に対する有効な治療法はなく、音声訓練等のリハビリテーション及び声帯の位置を移動する手術といった対症療法が中心となっております。
HGFの生物活性として線維化を抑制する抗線維化作用があるため、声帯瘢痕の治療にも活用できる可能性があると考えられます。当社では、京都大学医学部耳鼻咽喉科・頭頸部外科及び公益財団法人先端医療振興財団(現 公益財団法人神戸医療産業都市推進機構)との共同研究により、声帯瘢痕モデル動物の声帯内に組換えヒトHGFタンパク質を投与したところ、声帯機能の改善を認めました。そこで、医薬品の開発に必要な非臨床試験(声帯内投与における試験)を追加で実施し、臨床試験に開発ステージを進めました。声帯瘢痕患者を対象とした第Ⅰ/Ⅱ相試験結果の概要を下表に示します。
<表8:声帯瘢痕患者を対象とした第Ⅰ/Ⅱ相試験結果の概要>
当該試験の主要評価項目である安全性の確認について、重篤な副作用は認められず、忍容性も良好であることが示されました。また、副次評価項目において、改善傾向の見られる評価項目、評価時期についての知見が得られました。
これらの結果を基に、次相試験*の計画を策定しております。具体的には京都府立医科大学との協議の上、POCの取得を目的とするプラセボ対照二重盲検比較試験を想定し、複数の治験実施施設の選定、主要評価項目及び副次評価項目の設定を行い治験実施計画書骨子を作成しております。この計画書骨子を基に、2019年7月にPMDA*と事前面談を実施し、次相試験の協議を始めております。一方、治験実施費用の調達を目的として、各種公的補助金申請及び提携候補先の探索を継続しております。
声帯瘢痕においてHGFのPOCが確認された場合には、HGFの「抗線維化」作用に基づく創薬コンセプトそのものが示されることになり、声帯瘢痕のみならず他の線維化が原因となる慢性疾患(慢性腎不全、肝硬変、肺線維症等)への適応拡大の可能性が示されると考えております。
④ 急性腎障害
急性腎障害とは、腎臓の損傷、腎臓への血液供給不足、尿路の閉塞等により、数時間~数日という短い期間に急激に腎機能が低下する状態で、その結果、尿を介した老廃物の排泄ができなくなり、体内の水分や塩分量などを調節することができなくなる疾患です。重篤な場合には救急医療が必要になり、死亡に至る場合もあります。発症要因が多数あるため、原因を特定できない場合が多く、有効な治療法は確立されていません。HGFは腎臓の細胞に対して保護効果や増殖作用を示すことから、急性腎障害の治療薬となる可能性があると考えられます。当社は、米国の腎臓専門クリニック(Rogosin Institute)の協力を得て米国において臨床試験を行いました。ただし、第Ⅰ相試験においては、組換えヒトHGFタンパク質の安全性を確認することが目的であるため、容態の不安定な急性腎障害患者を対象とすることは不適切であると判断し、比較的容態が安定している慢性腎不全患者を対象として試験を実施いたしました。当該試験は、米国食品医薬品局(Food and Drug Administration:以下「FDA*」という。)により、必要性の高い新薬の審査を優先的に行う制度であるFast Track*の認定を受けて実施しております。試験結果の概要を下表に示します。
<表9:慢性腎不全患者を対象とした第Ⅰ相試験結果の概要>
当該試験において得られた情報を基に、次相試験として急性腎障害患者を対象とした臨床試験の計画を策定しております。また、国内での臨床試験実施を想定した第I相試験の追加試験(日本人での最大耐用量を確認する小規模試験)についても同時に策定しております。具体的には、急性腎障害を対象とした治験実施計画書の作成のため、医学専門家へのヒアリング、他社治験例の検討、バイオマーカー*調査等の検討を継続しております。しかしながら、次相試験は比較的大規模なプラセボ対照二重盲検比較試験になることが想定されており、現状では当社単独で神経系の治験と並行しての開発継続は難しいと判断し、製薬企業等と提携し、開発資金を確保した上で開発を進める方針としております。また現在、欧米において急性腎障害を対象とする競合品が第Ⅲ相試験の開発段階にあることから、その動向を注視しつつ、HGFの優位性を示すための開発を進めてまいります。
なお、静脈内投与は最も全身性に被験薬が到達するため、安全性の問題も発生しやすい投与経路になります。当該試験において安全性が確認されたことで、他の投与経路の開発を進める上で重要な知見が得られたと考えております。静脈内投与は様々な疾患に適応拡大しやすい投与経路であることから、急性腎障害に限らず、安全性及び有効性が効果的に確認できる疾患を策定しながら開発を進める方針です。
⑤ その他のパイプライン
当社は、国内外の大学や企業との共同研究において基礎研究を行い、新規パイプラインの強化を進めております。当社からは原薬あるいは治験薬の提供を行い、共同研究先にて有望なデータが得られた場合には成果を共有し、開発ステージを進める予定です。
<用語解説>
4 【関係会社の状況】
該当事項はありません。
5 【従業員の状況】
(1) 提出会社の状況
2020年10月31日現在
(注) 1.従業員数は就業人員(当社から社外への出向者を除き、社外から当社への出向者を含む。)であり、臨時雇用者数は従業員数の100分の10未満であるため、記載しておりません。
2.平均年間給与は、賞与及び基準外賃金を含んでおります。
3.当社は医薬品開発事業の単一セグメントであるため、セグメント別の従業員数の記載はしておりません。
4.最近日までの1年間において従業員が4名増加しております。主な理由は事業進捗に伴う業務拡大に対応するため期中採用が増加したことによるものであります。
(2) 労働組合の状況
労働組合は結成されておりませんが、労使関係は円満に推移しております。